
Il ruolo dell’entropia come descrittore
fenomenologico delle reazioni chimiche
Alessandro Motta
Dipartimento di Chimica, Università di Roma “La Sapienza”
e-mail: alessandro.motta@uniroma1.it
Indice
2. La prospettiva di Carnot in chiave moderna
3. Misura della variazione di entropia negli equilibri chimici
4. Il terzo principio della Termodinamica
Abstract. The presentation of the second law of thermodynamics in high school and early university teachings often leaves students with a sense of elusive abstraction, condensed into the expression “entropy measures disorder”. This could be avoided by treating entropy more specifically as a property derived from the analysis of heat exchanges between the system and the surroundings. In this contest, starting from a modern reinterpretation of Carnot’s work on the efficiency of heat engines, it would be helpful to delve into the analytical derivation of entropy associated with an isothermal expansion process, demonstrating how such a process is intimately related to the thermodynamic explanation of chemical equilibrium. Furthermore, it would be beneficial to explore the historical motivations that led to the formulation of the third principle to clarify its chemical importance in deriving reaction entropy.
Keywords: Carnot; entropia; equilibrio; terzo principio
Spiegare il concetto di entropia agli studenti che frequentano gli ultimi anni della scuola secondaria di secondo grado, come pure a quelli che si affacciano all’istruzione superiore, rappresenta un obiettivo arduo da raggiungere. Nella maggior parte dei casi, gli sforzi didattici atti a districare le innumerevoli sfaccettature dell’entropia vengono distillate in una risposta piuttosto vuota e demoralizzante: “l’entropia misura il disordine”. Questo, probabilmente, è dovuto alla difficoltà di coniugare gli aspetti fenomenologici legati al suo utilizzo pratico con la sua trattazione probabilistica a livello microscopico.
Di fatto, per capire a fondo l’entropia non solo dobbiamo sforzarci di immaginare cosa fanno atomi e molecole, ma per di più dobbiamo applicare formule che sono più vicine a quelle che regolano la probabilità nei giochi con i dadi, piuttosto che quelle che determinano la meccanica di Newton. Inoltre, la procedura per il calcolo quantitativo dell’entropia in esempi molto semplici, che spesso coinvolgono la distribuzione di qualche molecola tra due scatole, non è applicabile a casi macroscopici come transizioni di fase, o reazioni chimiche vere e proprie.
A conti fatti, il tentativo di derivare quantitativamente l’entropia dall’approccio microscopico rischia di rimanere scollegato dalla realtà, relegando questa funzione a quella cosa che misura il disordine. Da un tale punto di vista, la valutazione “sperimentale” dell’entropia in alcuni casi semplici, ma particolarmente importanti, aiuterebbe i ragazzi a rendere familiare e utile un concetto spesso limitato al mondo delle astrazioni.
In pratica, si dovrebbe rimarcare che l’entropia rappresenta uno degli strumenti (forse il più importante) presenti dentro la cassetta degli attrezzi del chimico.
In questa prospettiva, tre sono gli aspetti che verranno rimarcati qui di seguito. Il primo riguarda il contesto storico in cui è stata individuata l’entropia, sottolineando l’importanza didattica del lavoro di Carnot. Il secondo riguarda l’applicazione pratica di un calcolo di entropia associato all’espansione isoterma di un gas, semplice trasformazione, ma fondamentale per la comprensione degli equilibri chimici. Il terzo aspetto riguarda il contesto storico in cui è stato enunciato il terzo principio della Termodinamica per rimarcare come l’esigenza di trovare un modo di calcolare la variazione di entropia associata alle reazioni chimiche abbia condotto alla sua enunciazione.
2. La prospettiva di Carnot in chiave moderna
Se la direzione tracciata prevede l’utilizzo pratico dell’entropia, concepita come proprietà misurabile dei sistemi, allora il miglior punto di partenza è rappresentato da una rilettura in chiave moderna dello scritto principale di Sadi Carnot: “Réflexion sur la puissance motrice du feu et sur le machines propres as développer cette puissance” [1]. In maniera molto semplificata, la chiave moderna consiste nel tradurre la parola “calorico”, spesso usata da Carnot nel suo scritto, con la parola entropia. Infatti, sebbene il contesto teorico entro cui si muove Carnot sia concettualmente errato (la teoria del calorico non resisterà ancora a lungo dopo la morte di Carnot), le caratteristiche del calorico che Carnot utilizza per esplorare l’efficienza delle macchine termiche, rendono il calorico irresistibilmente simile al moderno concetto di entropia [2, 3].
2.1. L’entropia come proprietà
La prima fondamentale caratteristica che Carnot assegna al suo calorico, cioè alla nostra entropia, è che non si consuma durante la produzione di lavoro in condizioni ideali. Visto che la macchina è per definizione un dispositivo che funziona ciclicamente, significa che, alla fine di ogni ciclo di funzionamento (ideale), la quantità di entropia rimane invariata. Tanta ne entra e tanta ne esce. In chiave moderna diremmo che essa è una funzione di stato e, in quanto tale, possiamo considerarla come una proprietà qualificante dei sistemi: esattamente come la pressione, la temperatura e il volume, è possibile assegnare un valore di entropia ai sistemi.
Ritengo che questo sia un punto fondamentale che definisce come si vuole fare intendere l’entropia agli studenti, cioè come una proprietà qualificante dei sistemi. Qui, emerge il primo aspetto “misterioso” dell’entropia. Mentre le variabili come la pressione, il volume e la temperatura possono essere assegnate in maniera assoluta ai sistemi, non è possibile praticare una misura diretta di entropia dei sistemi, ma possiamo conoscere solo la sua variazione durante una trasformazione. Questa difficoltà, in realtà non è prerogativa dell’entropia, ma è condivisa con un altro concetto particolarmente sfuggente che si ritrova nello studio della Termodinamica, l’energia. Il fatto, forse, che rende l’entropia più sfuggente dell’energia è che essa si rende manifesta come proprietà dei sistemi solo se la trasformazione avviene in condizioni di idealità (reversibilità), mentre l’energia, o meglio la sua variazione, può essere misurata in qualsiasi tipo di trasformazione, semplicemente misurando il calore e il lavoro implicato nella trasformazione stessa. Ciò la rende più facilmente accessibile rispetto all’entropia.
Cosa intendere per condizioni di idealità è ben spiegato nello scritto di Carnot che introduce i concetti moderni di processo quasi statico, di reversibilità e di equilibrio. A questo proposito è necessario puntualizzare un aspetto che può indurre in equivoco. Se la trasformazione non avvenisse in condizioni di idealità o di reversibilità, non saremmo più in grado di fare una misura diretta della variazione di entropia. E se non è misurabile, allora esiste? Ovviamente sì. In questo caso, però, è necessario progettare un esperimento analogo in cui gli stati iniziali e finali del sistema siano gli stessi, ma il cammino con cui avviene la trasformazione sia ideale. Così facendo possiamo misurare la variazione di entropia e, visto che essa è una funzione di stato (cioè dipende solo dallo stato iniziale e dallo stato finale), tale variazione sarà la stessa della trasformazione irreversibile dove non eravamo in grado di misurarla direttamente.
2.2. Emerge una nuova funzione di stato
Una volta che sono state definite le condizioni sperimentali necessarie per poter fare una misura di variazione di entropia bisogna specificare come fare tale misura. Ciò viene dedotto in maniera lineare dal ragionamento di Carnot fatto per dedurre l’efficienza (η) massima di una macchina termica, cioè di una macchina in grado di compiere lavoro (L), grazie ad un flusso di calore (Qc) proveniente da una sorgente “calda” (Tc) verso una sorgente “fredda” (Tf), dove arriva parte del calore (Qf). Dalla definizione di efficienza (equazione 1):
[1]
e dalla constatazione che l’efficienza massima (ottenuta in condizioni di idealità) dipende solo dalla differenza di temperatura (equazione 2)
[2]
emerge che in condizioni di idealità la quantità (Q/T)rev, alla fine di un ciclo di funzionamento della macchina termica, è zero (equazione 3):
[3]
Quest’ultima relazione può essere interpretata dicendo che (Q/T)rev misura la variazione di una funzione di stato (una macchina funziona in modo ciclico e alla fine di ogni ciclo lo stato iniziale coincide con quello finale). Tale funzione di stato è stata chiamata entropia (S) e (Q/T)rev rappresenta proprio il modo di misurare la sua variazione (∆S). Sebbene queste considerazioni possano essere dedotte direttamente dalla lettura dello scritto di Carnot, il battesimo di questa proprietà risale al 1867, ad opera di Clausius [4]:
We might call S the transformational content of the body, just as we termed the magnitude U its thermal and ergonal content. […] I propose to call the magnitude S the entropy of the body, from the Greek word tropη, transformation. I have intentionally formed the word entropy so as to be as similar as possible to the word energy.
Clausius ricerca un’assonanza con la parola energia (U), per sottolineare la corresponsabilità delle due grandezze nel caratterizzare una trasformazione. Alla definizione di queste due grandezze corrispondono il primo e il secondo principio della Termodinamica. Il primo è un principio di conservazione e afferma che l’energia non si crea e non si distrugge (∆Uuni = 0), può solo trasformarsi come lavoro e calore, mentre il secondo possiamo considerarlo un principio di semi conservazione visto che in sostanza afferma che l’entropia dell’universo non si distrugge (∆Suni ≥ 0), e ci offre la possibilità di conoscere la freccia del tempo, cioè di prevedere da semplici calcoli se una trasformazione sarà spontanea oppure no.
3. Misura della variazione di entropia negli equilibri chimici
I concetti sinora esposti risultano già sufficienti per applicare le misurazioni di entropia alla vasta fenomenologia delle reazioni chimiche. Da questo punto di vista ritengo che ci sia un caso di particolare importanza. Mi riferisco all’entropia associata a un’espansione isoterma che è strettamente correlata alla trattazione dell’equilibrio chimico sia in fase gassosa che in soluzione.
3.1. Espansione isoterma di un gas: calcolo semplice, ma fondamentale
Questa trasformazione descrive banalmente cosa succede quando a un gas offriamo più spazio da occupare (e quando vi è un buon contatto termico con l’ambiente). Da un punto di vista chimico è una trasformazione molto importante perché avviene sempre durante una reazione in fase gassosa. Infatti, i prodotti formati (e i reagenti consumati) tendono a occupare tutto lo spazio a loro disposizione discostandosi dalla pressione standard. L’espansione isoterma quasi statica rappresenta il percorso reversibile da cui dedurre la variazione di entropia associabile poi a qualsiasi espansione durante una reazione chimica (Figura 1).
Figura 1. Calcolo dell’entropia associata a un’espansione isoterma di un gas
Per arrivare a questa formula è necessaria la conoscenza della legge dei gas ideali e prevede la risoluzione di un integrale notevole. Risulta essere, quindi, alla portata degli insegnamenti di chimica di base nella scuola secondaria e nei primi anni dei corsi universitari. La sua importanza risiede nel fatto che il semplice processo di espansione appena descritto è in grado di spiegare i processi di mescolamento e, in ultima analisi, l’equilibrio chimico, entrambi processi che, in condizioni ideali, si manifestano solo grazie a questo contributo entropico. Ci concentreremo adesso sul secondo processo, l’equilibrio chimico.
3.2. Equilibrio chimico
Capire l’equilibrio chimico da un punto di vista termodinamico significa capire perché una reazione non decorra sino a completamento, nonostante la variazione di energia libera di Gibbs calcolata in condizioni standard (∆G°reaz) sia negativa (o perché essa non decorra spontaneamente se tale variazione è positiva). Ricordiamo che la variazione di energia libera di Gibbs è intimamente legata alla variazione di entropia dell’universo secondo la semplice relazione ∆G = – Τ∆Suni.
Prendiamo il semplice esempio del diossido di azoto (NO2) che in fase gassosa è in equilibrio con la sua forma dimera (N2O4) e consideriamo la reazione di dissociazione del dimero (equazione 4).
[4] N2O4(g) ⇄ 2NO2 (g)
A temperatura ambiente il ∆G°reaz è positivo e, quindi la reazione non dovrebbe avvenire spontaneamente. Eppure, in fase gassosa si ritrovano entrambe le specie, il che significa che se partiamo solo dal dimero, la reazione un po’ decorre. Durante l’avanzamento di una reazione, sebbene la pressione totale rimanga sempre a 1 bar, i reagenti che stanno sparendo e i prodotti che si stanno formando si mescolano tra loro, ciascuno occupando tutto lo spazio a disposizione e abbassando, così, la propria pressione parziale sotto il valore di 1 bar (visto che la loro somma dà 1 bar). Possiamo immaginare il processo in avanzamento diviso in due stadi (Figura 2):
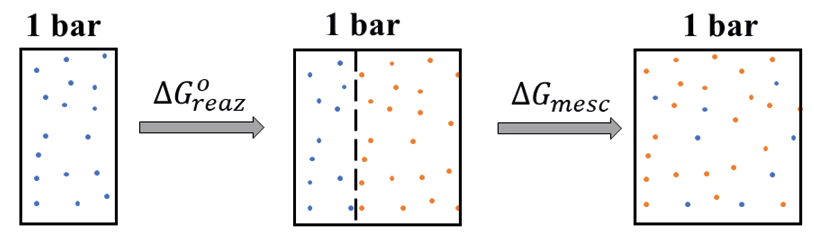
Figura 2. Contributi dell’energia libera durante il decorso della reazione che porta
alla decomposizione di N2O4 in NO2
Il primo stadio è descritto dal ∆G°reaz, mentre il secondo dal ∆Gmesc, che in condizioni ideali coincide con il –T∆Smesc, che può essere facilmente desunto dalla formula della espansione isoterma (equazione 5):
[5]
Se uniamo i due contributi otteniamo il valore totale di ∆Greaz al variare della composizione. Così facendo, si scopre che l’andamento di ∆Greaz tocca il suo minimo a una certa composizione tra i reagenti puri e i prodotti puri e che questo andamento è guidato esclusivamente dalla possibilità dei reagenti e dei prodotti di occupare tutto lo spazio a disposizione miscelandosi tra loro (sono gas dopo tutto). Quindi, la reazione decorre spinta dal contributo entropico del mescolamento fino a raggiungere la composizione di massima diminuzione dell’energia libera di Gibbs.

Figura 3. Andamento dell’energia libera durante il decorso di una reazione
che conduce all’equilibrio
A questo punto, se volessimo sintetizzare con una frase le motivazioni che spingono le reazioni chimiche a fermarsi a “metà strada”, cioè volessimo dare una interpretazione del concetto di equilibrio, potremmo dire che è tutta colpa della miscelazione tra reagenti e prodotti. Con questa semplice trattazione che prevede solo il calcolo di integrali notevoli risulterebbe chiara la derivazione termodinamica della legge di azione di massa e della relazione che esiste tra energia libera di Gibbs e costante di equilibrio. Concetti questi, che una volta introdotti accompagneranno gli studenti attraverso gli equilibri in fase gassosa, gli equilibri in soluzione, gli acidi e le basi, gli equilibri di solubilità e l’elettrochimica: praticamente metà del programma di un corso di chimica di base.
4. Il terzo principio della Termodinamica
Il terzo principio della Termodinamica non enuncia alcuna legge di conservazione e non introduce alcuna proprietà che determina le sorti dell’universo. Abitualmente nei corsi base viene enunciato come corollario che chiude il discorso sulla Termodinamica. In parole poco rigorose dice che l’entropia delle sostanze ben cristallizzate a zero Kelvin è nulla, ma può essere enunciato anche dicendo che non è possibile raggiungere lo zero Kelvin attraverso riduzioni finite di temperature. Sebbene, come sottolineato, non aggiunga ulteriori avanzamenti epistemologici rispetto ai primi due principi, il suo valore applicativo è di fondamentale importanza nella determinazione della spontaneità delle reazioni chimiche, come dimostra il contesto storico in cui è stato enunciato. Prima di tutto capiamone l’importanza strumentale.
Un metodo sperimentale generale per acquisire informazioni sulla variazione di energia libera di Gibbs associata ad una reazione chimica (∆Greaz), è quello di misurare il calore di reazione a pressione costante (qp), che ci conduce alla conoscenza del contributo entalpico (∆Hreaz), e il calore di reazione in condizioni di reversibilità (qrev), che ci conduce alla conoscenza del contributo entropico (∆Sreaz = qrev/T). I due contributi sono legati al ∆Greaz dalla nota relazione (equazione 6):
[6] ∆G = ∆H – T∆S
Mentre il calore di reazione è facilmente accessibile, trovare delle condizioni di reversibilità per le reazioni chimiche, dalle quali dedurre il contributo entropico, non è così banale. Alcune reazioni possono essere fatte avvenire in una cella elettrochimica e, in tal caso, da misure di potenziale (∆E) si può ricavare direttamente il ∆Greaz attraverso la legge di Nernst (∆G = −nF∆E). In altri casi, il ∆Greaz può essere ricavato dalle condizioni di equilibrio (∆G°reaz = −RTlnK). In molti altri casi (si pensi, ad esempio, alle combustioni) la valutazione del ∆Greaz deve passare dalla stima del ∆Sreaz, quando questo non è suscettibile di misura diretta. Se non può essere valutato direttamente, possiamo pensare di farlo indirettamente. Nel caso dell’entalpia, per esempio, era possibile esprimere il ∆Hreaz introducendo il concetto di entalpia di formazione delle singole sostanze coinvolte nella reazione, ma per il ∆Sreaz il problema di trovare le condizioni di reversibilità riguarda ovviamente anche l’entropia di formazione delle singole sostanze. Il problema sarebbe semplicemente spostato, ma non risolto. È qui che ci viene incontro il terzo principio della Termodinamica. All’inizio del 1900 si sapeva calcolare l’incremento di entropia associata al riscaldamento di una sostanza (equazione 7):
Questa formula, partendo dallo zero Kelvin, poteva restituire il valore assoluto dell’entropia di una sostanza (ST) a una data temperatura (equazione 8)
solo se si riusciva a stimare l’entropia di una sostanza allo zero kelvin (S0). Mentre il termine integrale era calcolabile, il termine S0 doveva essere estrapolato. Immaginate di voler risalire all’altezza di un individuo di 15 anni sapendo che mediamente cresce 10 cm all’anno. Ovviamente non è possibile, se non si conosce anche la sua altezza al momento del parto. Per la determinazione dell’entropia assoluta sussisteva lo stesso problema: si poteva conoscere l’incremento di entropia con la temperatura (il termine integrale), ma non si conosceva la temperatura “nativa” (S0). La sfida per la determinazione S0 fu raccolta da diversi ricercatori, tra cui Le Chatelier [5] e Lewis [6]. Alla fine, nel 1906 Nernst [7] enunciò quello che, con qualche modifica, è conosciuto come terzo principio della Termodinamica assegnando per tutte le sostanze S0 = 0. La conoscenza dell’entropia assoluta ci permette di conoscere l’entropia di reazione (equazione 9) dalla conoscenza del contenuto entropico di ciascuna specie presente tra i prodotti (Sp) e tra i reagenti (Sr)
e in ultima analisi ci permette di conoscere il ∆Greaz potenzialmente di tutte le reazioni chimiche. Se si contestualizza storicamente l’enunciazione del terzo principio e le necessità pratiche che hanno condotto alla sua formulazione, risulta evidente la sua importanza nell’ambito delle reazioni chimiche.
Ancora due commenti sui concetti di energia ed entropia. Siamo abituati a parlare di energia in contesti familiari e quotidiani, mentre il concetto di entropia appare molto più astratto e sfuggente, ma dalla Termodinamica ci arriva un messaggio differente. Riguardo l’energia, nonostante sia una proprietà dei sistemi (sia considerata come energia interna che come entalpia) non siamo in grado di attribuirne un valore a un determinato stato, ma siamo capaci solo di conoscerne la variazione durante una trasformazione che porta da uno stato iniziale ad uno finale. Inoltre, dietro la parola energia si nasconde un mondo variegato che comprende tanti tipi di manifestazione dell’energia stessa: energia potenziale, energia cinetica, lavoro e calore. D’altro canto, parlando di entropia, non solo abbiamo un concetto univoco (l’entropia non si manifesta sotto diverse forme), ma è possibile assegnare un valore di entropia ai sistemi (almeno in certe circostanze) oltre che calcolarne la variazione durante una trasformazione. Di sicuro l’entropia è più sfuggente dell’energia, visto che si manifesta solo in condizioni di reversibilità, ma se ci limitiamo al suo uso strumentale ci rendiamo conto per certi aspetti che è un concetto meno problematico dell’energia. Infine, è possibile dare un volto “microscopico” all’entropia, qualificandola come misura del grado di dispersione dell’energia delle molecole. Gli esempi per estrinsecare questo concetto sono molteplici, mi limiterò a elencare quelli più strettamente connessi agli argomenti trattati nelle sezioni precedenti. Un primo esempio è proprio l’espansione isoterma di un gas descritta nella sezione 3, in cui la dispersione deriva dalla possibilità delle molecole di muoversi in uno spazio più grande. Un secondo esempio passa dal confronto delle distribuzioni di Maxell-Boltzmann delle energie cinetiche di una sostanza a diverse temperature. In questo caso la dispersione è rappresentata proprio dall’allargamento della distribuzione all’aumentare della temperatura e spiega l’aumento dell’entropia delle molecole all’aumentare della temperatura. Infine, un terzo esempio si incontra nei passaggi di stato dove la dispersione si manifesta come aumento dei gradi di libertà delle molecole passando dal solido (le molecole possono solo vibrare) al liquido (le molecole ruotano e si spostano, ancora parzialmente vincolate) e al gas (totale libertà). Questi due ultimi esempi sono collegati alla misura dell’entropia assoluta delle sostanze come descritto nella sezione 4.
[1] S. Carnot, Réflexions sur la puissance motrice du feu et sur les machines propres à développer cette puissance, Bachelier, Paris, 1824.
[2] H. Yu, Y.-X. Liu, G. Falk, Entropy, a resurrection of caloric-a look at the history of thermodynamics, Eur. J. Phys., 1985, 6, 108.
[3] H. L. Callendar, The caloric theory of heat and Carnot’s principle, Proc. Phys. Soc. London, 1910, 23, 153–189.
[4] R. Clausius, The mechanical theory of Heat, 1867.
[5] H. Le Chatelier, Recherches expérimentales et théoriques sur les équilibres chimiques, dunod, 1888.
[6] G. N. Lewis, A general equation for free energy and physico-chemical equilibrium, and its application, PhD dissertation, Harvard University, 1899.
[7] W. Nernst, Über die berechnung chemischer gleichgewichte aus thermischen messungen, Nachrichten von der gesellschaft der wissenschaften zu Göttingen, Math. Klasse, 1906 (n.d.) 1–40.